
Han sido semanas muy ocupadas en los últimos días en el reumaverso, con mucha consulta, trabajos pendientes, tamizaje, aleatorización, notas y claro, una fila interminable de pacientes, lo cual me distrajo un poco del foco actual en el que me encontraba, si bien es difícil para mi escribir, la inspiración me llega como estímulos de serotonina cada vez que veo a los amigos de la cohorte nounish, contentos y emocionados.
La historia de hoy, de cómo llegó la inspiración es corta, pero curiosa. En la red verde, la cual normalmente ignoro, si no es por trabajo o para contestar dudas, me sorprendió una burbuja de más de 100 mensajes, lo cual es poco frecuente, salvo que exista algún meme o algún médico haciendo cosas cuestionables, pero esta ocasión era un caso clínico, de una doctora que aprecio mucho, expresando sus dudas. Como miel, todos nos volcamos a platicar y a decidir cuáles eran las aproximaciones diagnósticas, lo que denota nuestro comité de expertos y sobre todo experiencia que hoy nos caracteriza, el tema es histiocitosis.

¿Qué es la histiocitosis?
La palabra suena bonita, tiene un inicio griego , el cual se traduce como “célula de tejido”. El artículo al que logré remontarme, es de 1925, una descripción hecha por el Dr. Frank A. McJunkin, en donde describe “la presencia de leucocitos granulares polimorfonucleares circulantes”, las cuales al parecer, tenían un origen en la médula ósea, pero viajaban a otros tejidos como el bazo, lo que dificulta en ese tiempo su extracción. Al realizar tinciones especiales el Dr. Ehrlich en 1879, como la tinción de 3 ácidos, descubrió leucocitos mononucleares, inicialmente clasificados como monocitos, que se convierten posteriormente en células polimorfonucleares, en la circulación, lo que les otorgó el nombre de “leucocitos gigantes mononucleares” o “leucocito transicional”.
Los hallazgos, fueron la idea que sembró la curiosidad en Mallory, Aschoff, Kiyono y el grupo de Sabin, inicialmente clasificados como “leucocitos endoteliales”, “leucocitos tisulares”, “leucocitos de migración”, por el comportamiento in vitro que se observaba, dado que las células recién descubiertas tenían un comportamiento curioso, parecían células adheridas a los vasos sanguíneos, las cuales se hacían libres para convertirse en fagocitos o migrar por la composición del vaso.
Al ser estimuladas, con antígenos, éstas células adquieren una forma en roseta (alrededor del agente externo), al ser expuestas al carbón, experimentos que fueron inicialmente realizados en conejos, las cuales posteriormente fueron estimuladas en tejido humano, con bencidina observadas por el Dr. Charlton y McJunkin, logrando aislar al fagocito endotelial, fácilmente diferenciado del monocito “normal” que no reacciona con bencidina.
Hoy sabemos que estas células tienen un origen desde las células dendríticas o de los macrófagos. La primera categoría descrita, fue publicada en 1987 por El Working Group of the Histiocyte Society (HS), la cual las divide en tres grupos Célula de Langerhans, Célula no Langerhans y los histiocitos malignos. La división actual se promueve en cinco categorías, Grupo C (cutáneo), Grupo H (histiocitosis), L (Langerhans, Mixto), Grupo M (Malignidad primaria) R (esporádico).
Los histiocitos derivan del sistema fagocítico mononuclear, en donde encontramos a las células dendríticas con forma de estrella, las cuales tienen un complejo principal de histocompatibilidad para la presentación antigénica, complejos proteicos y estimulación de linfocitos; se dividen en dos grupos, las mieloides y las plasmocitoides. Las mieloides se subdividen en CD141 (mDC1), y CD1c (mDC2). Las células de Langerhans son un tipo de célula dendrítica presente en la epidermis, mucosa epitelio bronquial y detección de CD1a con gránulos de Birbeck, aunque en la actualidad se definen por la presencia inmunohistoquímica de CD1a y CD207.
Algunos aspectos genéticos
Con el avance en el entendimiento del genoma, se ha identificado que ciertas mutaciones y traslocaciones en el gen BRAF p.V600E en pacientes con histiocitosis de Langerhans ofrecen un contexto fisiopatológico que permite comprender mejor la enfermedad, esta alteración genética específica, resulta en la sustitución del ácido glutámico por valina en la posición 600 del gen BRAF. Esta mutación provoca una activación constitutiva de la proteína BRAF, lo que conduce a la activación continua de la vía de señalización oncogénica MAPK (mitogen-activated protein kinase).
La vía MAPK es crucial para la regulación de diversas funciones celulares, incluyendo la proliferación, diferenciación, y supervivencia celular. Cuando esta vía está desregulada debido a mutaciones como la BRAF p.V600E, puede resultar en un crecimiento celular anormal y contribuir al desarrollo de enfermedades como la histiocitosis de Langerhans.
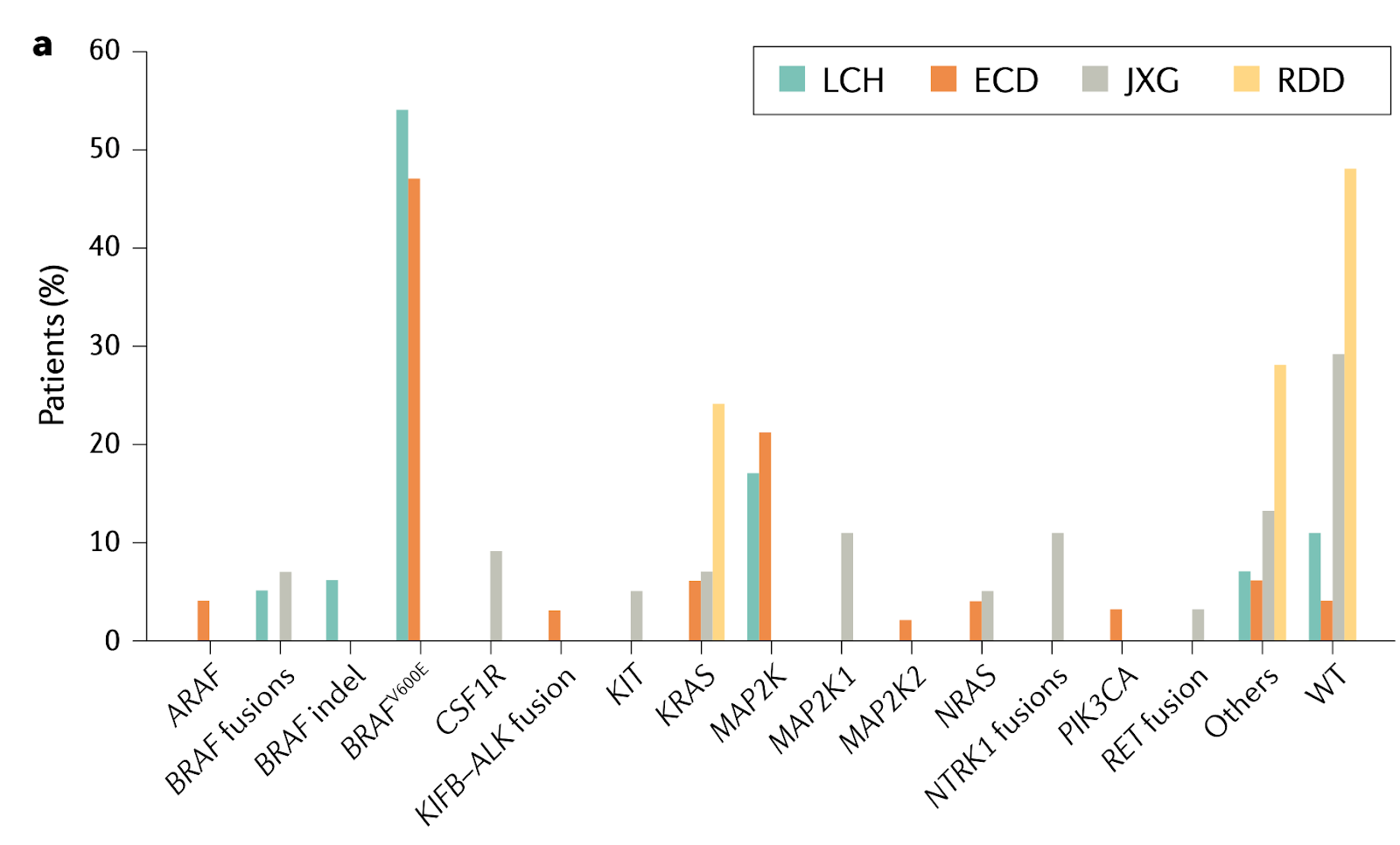
Esta misma vía de señalización MAPK se ha observado activada en otras mutaciones oncogénicas, incluyendo las mutaciones en los genes PIK3CA, PICK1, PICK3R2, ALK y NTRK1. Por ejemplo, el gen PIK3CA codifica una subunidad de la fosfatidilinositol-3-quinasa (PI3K), que participa en la vía PI3K/AKT/mTOR, una ruta paralela y a veces interconectada con la vía MAPK, que también está implicada en la regulación del crecimiento y la supervivencia celular. Las mutaciones en PIK3CA pueden llevar a la activación anormal de esta ruta, contribuyendo a procesos similares de desregulación celular.
De manera similar, las alteraciones en los genes ALK y NTRK1 están relacionadas con la activación de tirosina cinasas, que son enzimas cruciales en las vías de señalización celular. Estas mutaciones pueden resultar en la activación aberrante de las vías de señalización que promueven el crecimiento y la supervivencia celular, conduciendo a condiciones patológicas.
Sin embargo, la asociación con oncogenes no implica necesariamente que la histiocitosis de Langerhans sea una condición maligna. Las características que definen la malignidad incluyen mutaciones en oncogenes, resistencia a la apoptosis, autosuficiencia en señales de supervivencia, invasión de tejidos y metástasis. En cambio, en la histiocitosis no hay una replicación celular acelerada, y la mayoría de las asociaciones son con genes relacionados con la senescencia.
Las formas más graves de esta patología están estrechamente relacionadas con diversas inmunodeficiencias. Entre las mutaciones genéticas más comunes que predisponen a una mayor severidad de la enfermedad se encuentran aquellas en los genes MAGT1, ITK, CD27, CTPS1, MST1, DOCK8, DOCK2, ORAI1 y STIM1. Estas mutaciones afectan componentes críticos del sistema inmunológico, resultando en una respuesta inmunitaria comprometida. Por ejemplo, la mutación en MAGT1 puede alterar la homeostasis del magnesio y afectar la función de las células T, mientras que las mutaciones en ITK están asociadas con defectos en la señalización de los receptores de células T, cruciales para una respuesta inmunitaria efectiva.
Además, la deficiencia de coronina 1A, una proteína importante para la motilidad y la supervivencia de las células T, también se ha identificado como un factor que exacerba la severidad de la patología. La falta de coronina 1A puede llevar a una disfunción grave en la migración y supervivencia de los linfocitos, contribuyendo así a una inmunodeficiencia significativa.
Otros factores que pueden desencadenar fenómenos agresivos en esta enfermedad incluyen diversos trastornos reumatológicos. La enfermedad de Still, el lupus eritematoso sistémico (LES) y la artritis idiopática juvenil son condiciones autoinmunes que pueden agravar la patología. En la enfermedad de Still, por ejemplo, la inflamación sistémica severa puede desencadenar complicaciones adicionales, como pancreatitis, insuficiencia respiratoria aguda y disfunción del sistema nervioso central. El lupus eritematoso sistémico, conocido por su amplia gama de manifestaciones clínicas, puede provocar daño multisistémico, desenfrenado con citopenia profunda y hemorragia alveolar. De igual manera, la artritis idiopática juvenil, con su inflamación articular crónica, puede contribuir a una mayor complejidad clínica y severidad de la enfermedad.
Además de los trastornos reumatológicos, las infecciones virales también juegan un papel crucial en la exacerbación de la patología. El virus de Epstein-Barr (EBV), en particular, ha sido identificado como un desencadenante significativo. Este virus, que se asocia con diversas enfermedades autoinmunes y linfoproliferativas, puede reactivar y complicar la condición existente, induciendo una respuesta inmunitaria aberrante que agrava los síntomas y la progresión de la enfermedad.
¿Qué implica este tipo de padecimientos?
La histiocitosis de Langerhans conlleva un grupo amplio de enfermedades de predominio en adultos y niños, desde lesiones localizadas con resolución espontánea, hasta daño orgánico que pone en peligro la vida. Pueden presentarse en los huesos en el 80% de los casos, piel 22%, glándula pineal 25%, hígado, bazo, sistema hematopoyético o los pulmones 15%, ganglios linfáticos 5-10% y sistema nervioso central 2-4%.
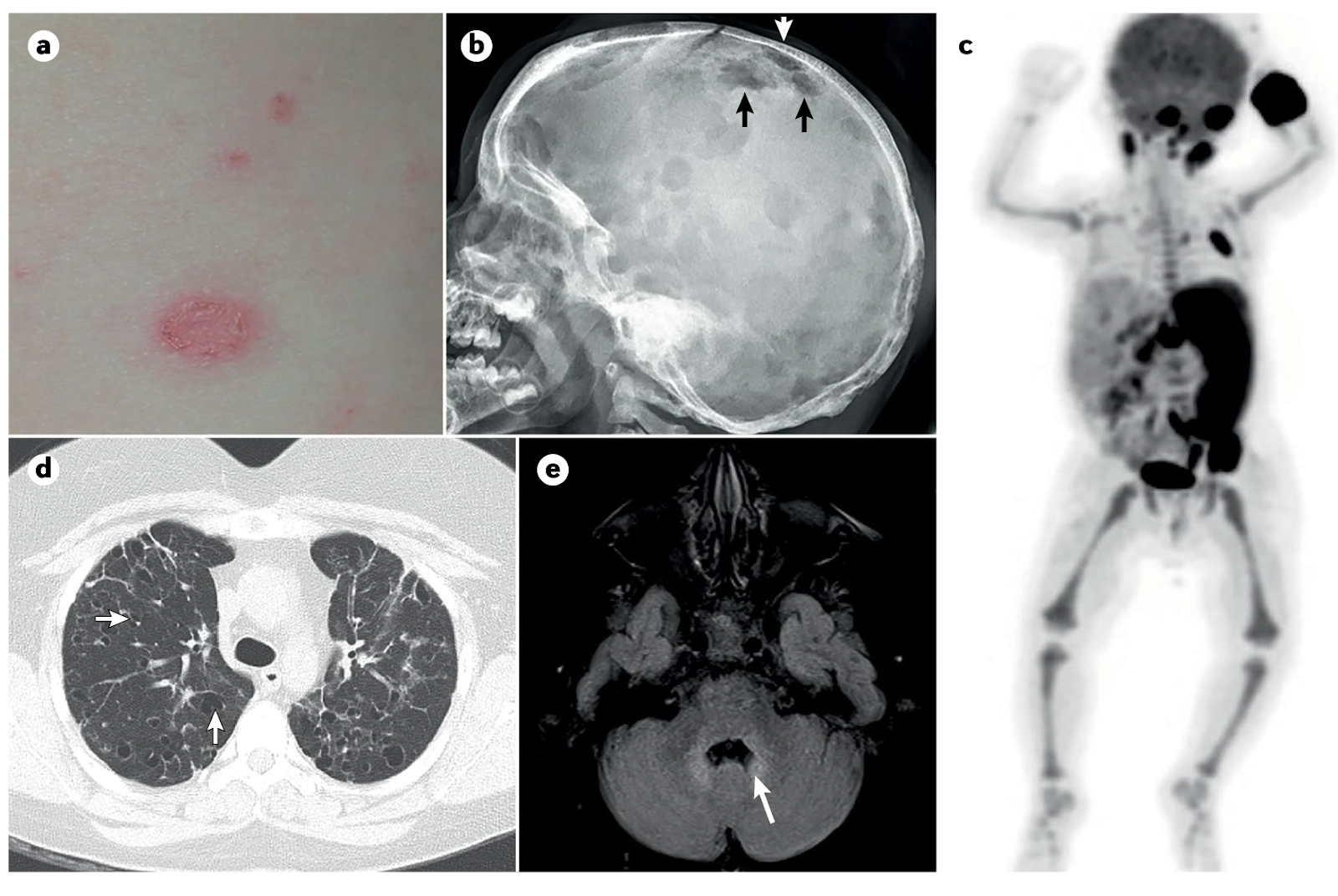
Tenemos muchas variantes que afectan grupos de edad y órganos diferentes, como por ejemplo:
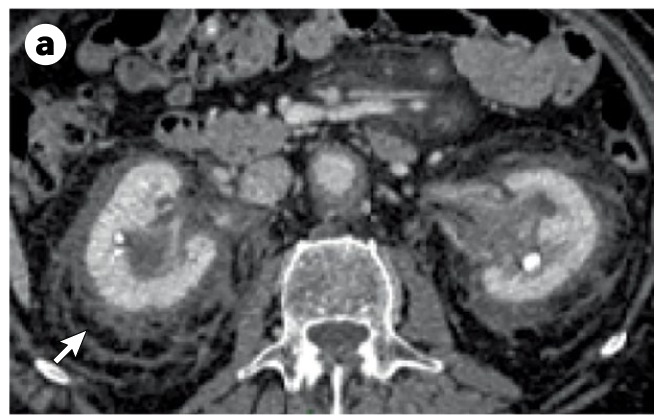
El síndrome de Erdheim-Chester, el cual aparece entre los 55 y 60 años, de predominio en hombres 3:1, padecimiento que afecta en su mayoría el esqueleto en un 95% de los casos, causado osteosclerosis de la diáfisis y metáfisis, 50% presenta afección cardíaca, fibrosis retroperitoneal, alrededor de los riñones y ureter. Estos pacientes tienen positividad a CD68 y CD163, ocasionalmente a S100 con emperipolesis (eritrocitos, células plasmáticas y linfocitos fagocitados por histiocitos).

El síndrome de Rosai-Dorfman-Destombes, inicialmente descrito por Destombes en 1965, es más frecuente en niños y adultos jóvenes,consiste en ganglios en la región cervical, asociados con fiebre, sudoración nocturna, fatiga y pérdida de peso, también se pueden encontrar nódulos mediastinales, inguinales y retroperitoneales, en el 43% de los casos también se han descrito lesiones en piel, cavidad nasal, hueso y retro orbitario, para el diagnóstico de este padecimiento es necesaria la tinción inmunohistoquímica de la emperipolesis S100+, fascina+, CD68+, CD14+, HLA-DR+, macrófagos CD163+ conCD1a o CD207; ocasionalmente pueden presentar niveles muy alto de IgG4, lo cual hace difícil el diagnóstico diferencial con estas enfermedades.
Xantogranuloma juvenil, es un padecimiento que se presenta en menores de 4 años, con una lesión única o múltiple de color naranja rojiza, de características papulares, las cuales tiene una resolución espontánea. Ocasionalmente pueden existir padecimientos sistémicos que afectan el hígado, bazo, riñón, cerebro, hueso, el cual se vuelve progresivo y ocasionalmente fatal.
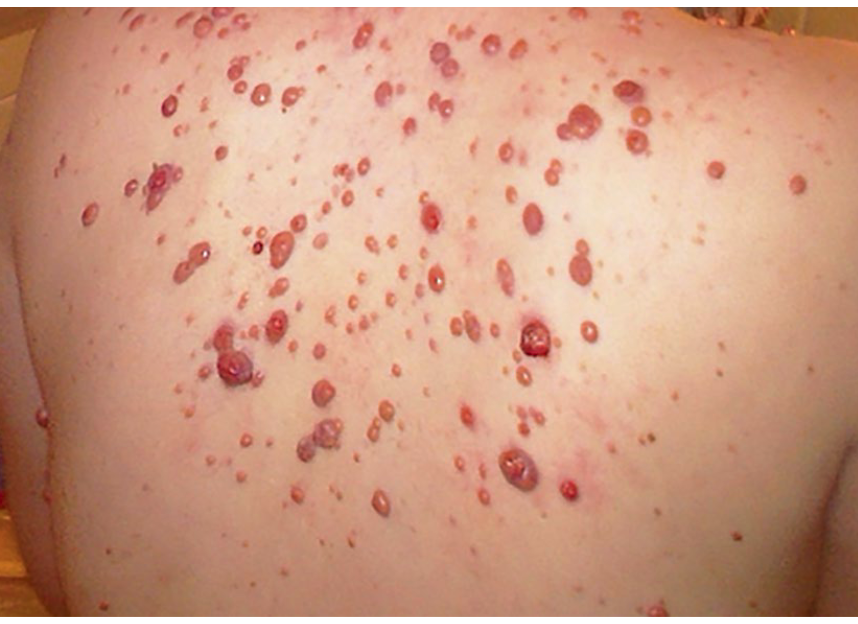
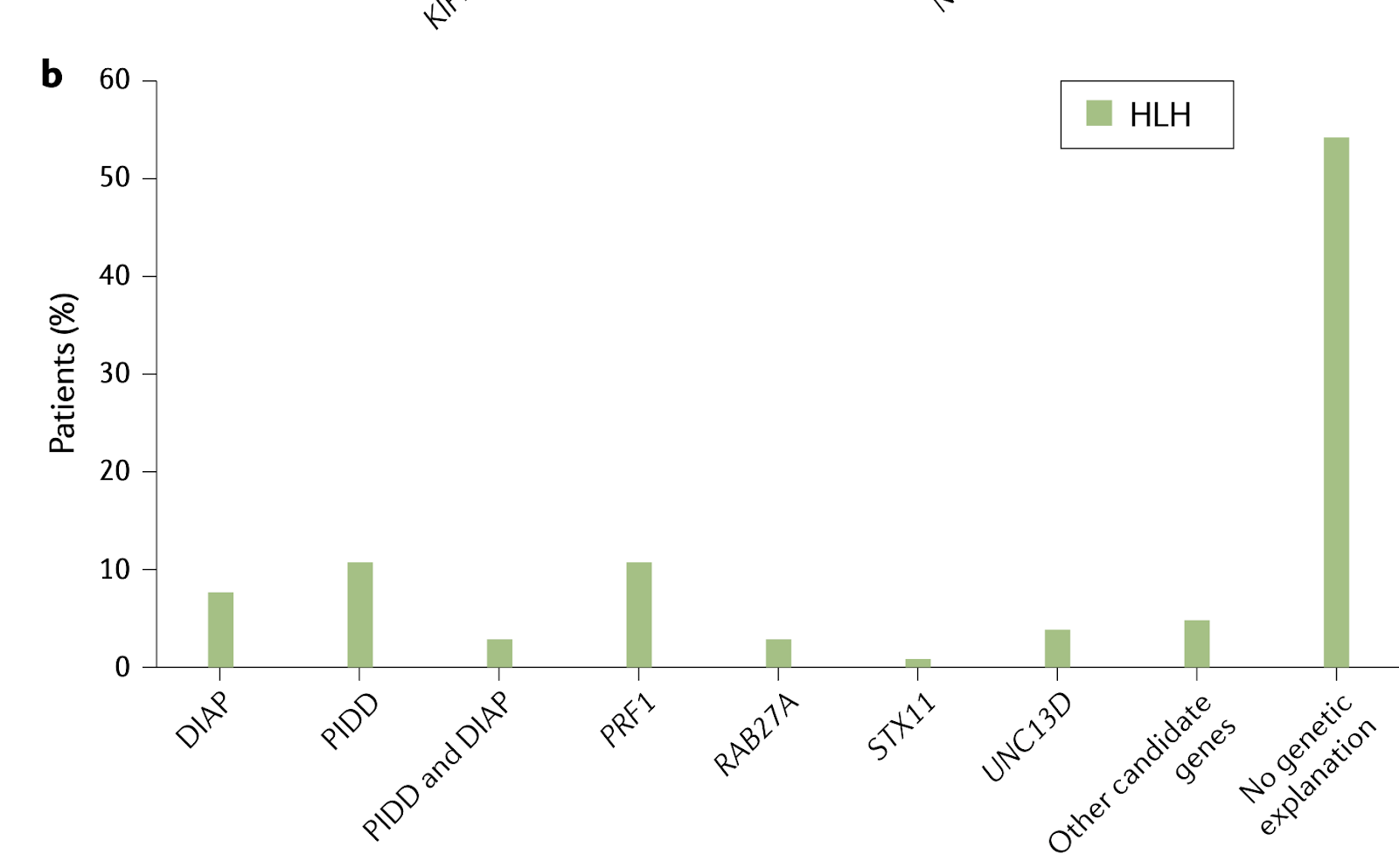
Linfohistiocitosis Hemofagocítica, es un padecimiento grave, ocasionalmente fatal, el cual es causado por una regulación inapropiada de las citocinas, causando una hiperactivación de macrófagos. La mayoría de los casos están asociados con un desorden en la regulación del inflamasoma, lo cual permite una inflamación descontrolada.
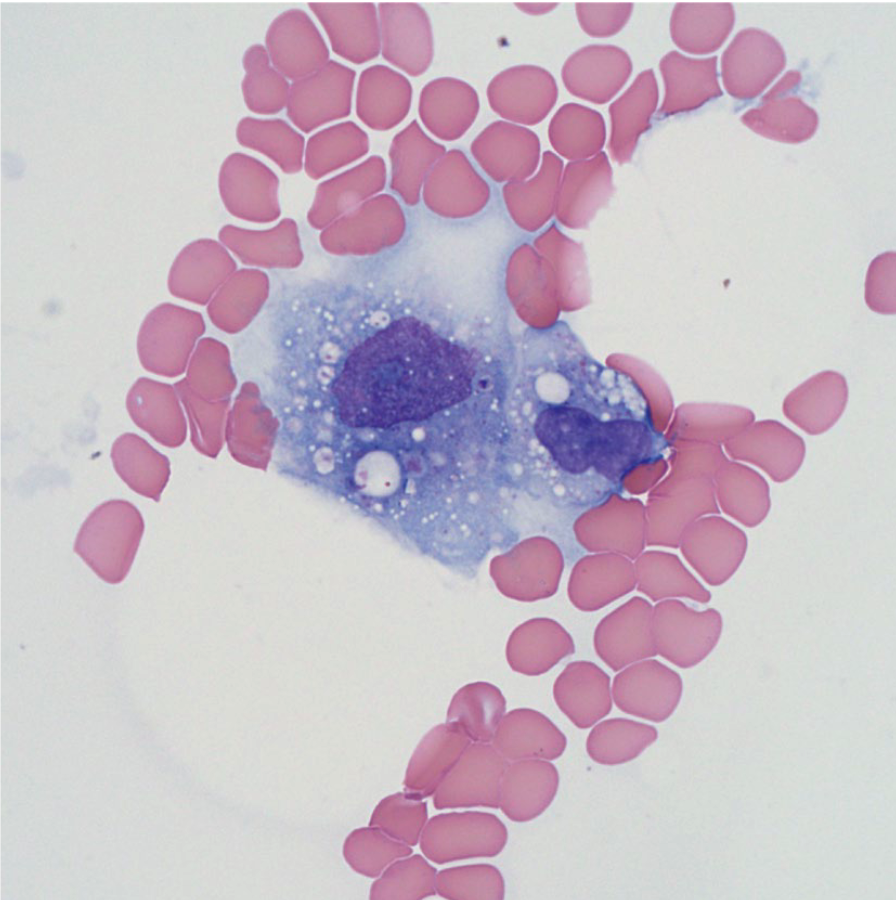
Artículo a revisar
El tratamiento de las histiocitosis es complejo, hasta el momento solo tenemos solo el tratamiento con trasplante de células hematopoyéticas troncales, sin esta terapia, los pacientes están en riesgo de recaída, daño multiorgánico o nula respuesta. En cuanto al tratamiento quimioterapéutico puede provocar mielotoxicidad, infecciones frecuentes, y morbilidad. La propuesta es darle espacio a Emapalumab, un anticuerpo monoclonal que neutraliza el interferón gamma, el cual ha demostrado mejoría en los parámetros de laboratorio y clínica.
Nuestro estudio fue diseñado por los investigadores de REAL-HLH, en un ambiente retrospectivo, no intervencional, en 33 hospitales de estados unidos, seleccionando a los pacientes que hayan recibido emapalumab entre 2018 y 2021.
Se seleccionaron pacientes con mutaciones genéticas relacionadas con histiocitosis, cumpliendo por lo menos 5 criterios de 8 de la clasificación HLH-2004, historia familiar del HLH. El objetivo primario del protocolo fue la descripción demográfica, características clínicas y tratamiento de los pacientes con pHLH tratados con epalumab, razón del inicio, patrón de dosificación, duración de tratamiento, otras terapias iniciadas con el epalumab y uso de glucocorticoides en las primeras ocho semanas.
En análisis estadísticos dividió a los pacientes a partir de los 12 años, se reportaron curvas de supervivencia de Kaplan-Meier.
Resultados: Encontraron 105 pacientes, 98 de ellos elegibles, al realizar el tamizaje con el HLH-2004 46 (46.9%), de los pacientes cumplían los criterios del protocolo para su inclusión, tiempo de diagnóstico de 1 año, la mayoría eran niños menores de 2 años, varones. Las mutaciones más frecuentemente encontradas fueron la PRF1 (n = 15; 37.5%), UNC13D (n = 10; 25%), y LYST (n = 5; 12.5%). La edad media de inicio de la terapia con epalumab fue de un año, la mayoría de los pacientes estaban graves, ya sea en terapia intensiva o con soporte vital.
El tiempo medio de normalización de los parámetros de laboratorio fue de 7 días, entre los que se incluyeron el fibrinógeno, conteo absoluto de neutrófilos, plaquetas, coneto absoluto de linfocitos y alanin aminotransferasa.
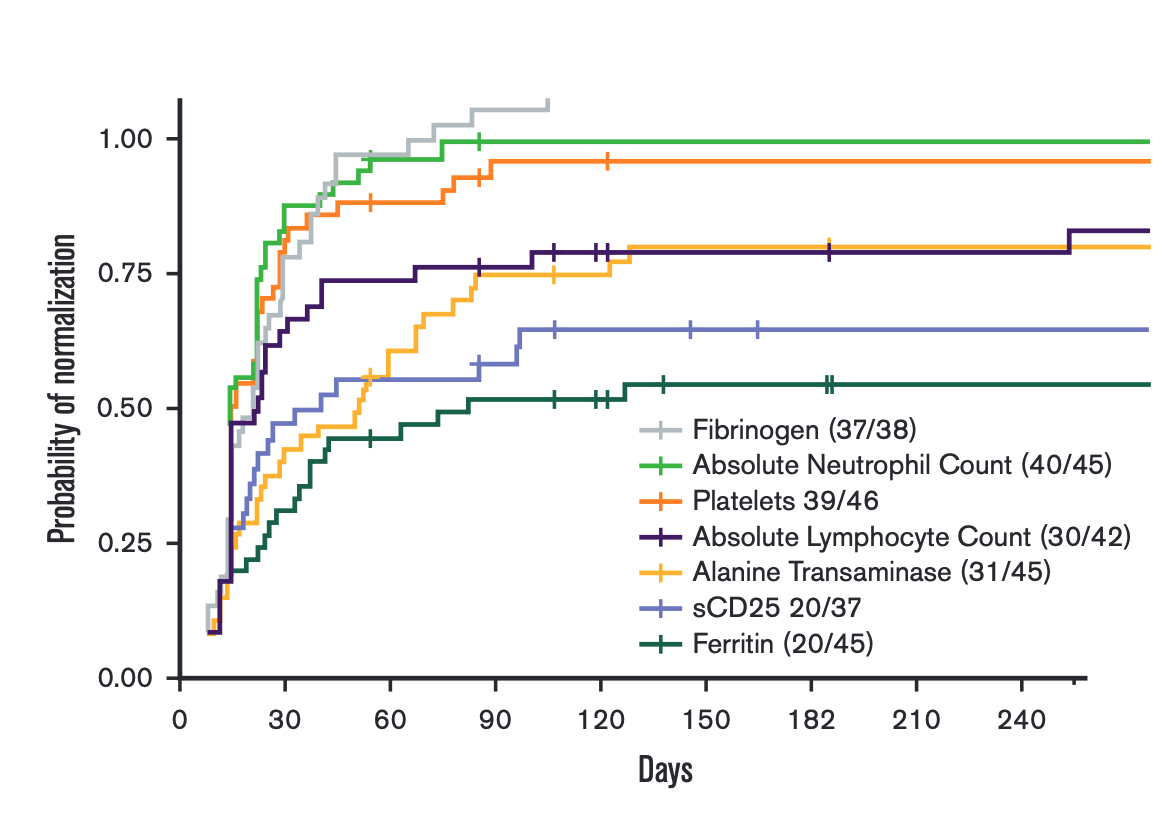
La supervivencia de los pacientes alcanzó el 73.9% al año, lo que permitió una mejoría en la mortalidad para los pacientes que recibieron epalumab antes y después del trasplante.

Con base en el análisis del estudio, sabemos que el título es coherente con lo que se describe durante el protocolo, el diseño del estudio es bueno, debido a que la infrecuencia del padecimiento, el costo de la terapia y el riesgo alto de mortalidad, no permite por cuestiones éticas el uso de terapias sin tratamiento estándar, lo que más adelante permitirá usar otros medicamentos en comparación con epalumab, si se replican estos estudios. En realidad, los datos son alentadores lo que motiva a su vigilancia y posibilidades de tratamiento para nuestros pacientes.
Un vistazo al futuro
Las histiocitosis son enfermedades infrecuentes, complejas, ocasionalmente graves, incluso con posibilidad de poner en peligro la vida de los pacientes. Los aspectos genéticos y las alteraciones que describimos aquí son un pequeño esbozo de lo que podemos encontrar y se me hizo un caso en extremo interesante. Hasta el momento no sabemos que tiene el paciente de uno de mis compañeros, pero muchos cerebros piensan mejor que uno. Les dejo con la ciencia, en esta ocasión compleja y nos deja pensativos. Vayamos aprendiendo juntos, dejando la ciencia sinérgica en nuestro escaparate cada semana.
Nos vemos la siguiente semana :D.
Gobernanza en Ciencia
🏛AthenaDAO
Snapshot: ✅ ADP-006: Establishing a Data Working Group (Pasada). Sin cambios
💛 Vita DAO:
Actualmente en discusión
Foro 😃
VDP-152 [Funding] Project TransFidelity (Avtiva)
VDP-149 Clarification and Maintenance of VitaDAO Governance and Proposals (activa)
VDP-148 Blueprint for Sustainable Growth: Operationalizing VitaDAO’s New Mandate (cerrada)
Snapshot ✅: VDP-146 [Funding] The development of oligonucleotide drugs for healthspan (Pasada)
Snapshot ✅: VDP-148 Blueprint for Sustainable Growth: Operationalizing VitaDAO’s New Mandate (Pasada)
❄CryoDAO:
Snapshot ✅ CRYO-18: CryoDAO <> Molecule Partnership (Pasada)
🧠CerebrumDAO
Snapshot: ✅ CDP-7: Bridge NEURON to Base and Create a Liquidity Pool on Aerodrome Finance (Pasada).
🌳ValleyDAO
Snapshot: VIP-10: Continuation of GROW/ETH Pool Incentives on Base
💈HairDAO

 34
34
BioProtocol


@scienceman2023 of Pfizer Ventures discusses the power of onchain IP & @vita_dao
Are Biotech & Healthcare the next frontier for crypto's real-world adoption?
Primer artículo pre publicado de HairDAO: https://www.biorxiv.org/content/10.1101/2024.06.11.598522v1

🤑Funding:



https://www.molecule.xyz/researchers
Divulgación. De vez en cuando podemos añadir enlaces en este boletín a productos que utilizamos o poseemos. Podemos recibir una comisión si haces hace una compra a través de uno de estos enlaces. Además, los redactores de Bankless poseen criptoactivos. Vea nuestras divulgaciones de inversión aquí y las de Nación Bankless aquí. Recuerda que BanklessDAO-Nación Bankless y BanklessHQ son entidades independientes.
🪁 Tenemos una alianza con nuestros amigos de Espacio Cripto que ahora han lanzado -Bando, una herramienta para el off y on Ramp seguro, tan fácil como un SPEI.

🧑💻 No olvides leer Octant y su funcionamiento
🦄 Únete: a nuestro telegram para tener buenas charlas, saber de noticias, estar enterado de todo lo que estamos construyendo para la comunidad hispana.
💳 Quieres utilizar BANK y NOGS como puntos de recompensa para tu DAO favorita. Actualmente bCARD esta en alfa y en las próximas semanas tendremos el beta disponible en Google Pay y Apple pay.
🔓 Staking4, es una plataforma que creamos para ti, para que vayas a hacer staking de tu ETH en Meta Pool de una forma sencilla y eficaz. Sumérgete a las NB.

¿Quieres conocer todo lo que pasa en el ecosistema cripto y web3 día a día? Pues bien, puedes suscribirte a Bankless para estar al tanto de todo lo que pasa y invita a tus amigos para que no se lo pierdan 🚀
Gracias por leer Nación Bankless! Suscríbete para tener acceso al contenido premium de Bankless sin costo y continuar apoyando nuestro trabajo.

